Clone Library Dereplicator 2.7
- Developer Link 1 (non https DNA Baser Setup.exe)
- Developer Link 2 (non https DNA Baser Setup.exe)
- Developer Link 3 (non https)
- Download3k US (ver 2.62, DNA Baser Setup.exe)
- Download3k EU (ver 2.62, DNA Baser Setup.exe)
MD5: e1764bef6be7b10b45085041ead79baf
All files are original. Download3K does not repack or modify downloads in any way. Check MD5 for confirmation.
Developer Description
"www.DnaBaser.com - Find duplicate DNA sequences and remove them."
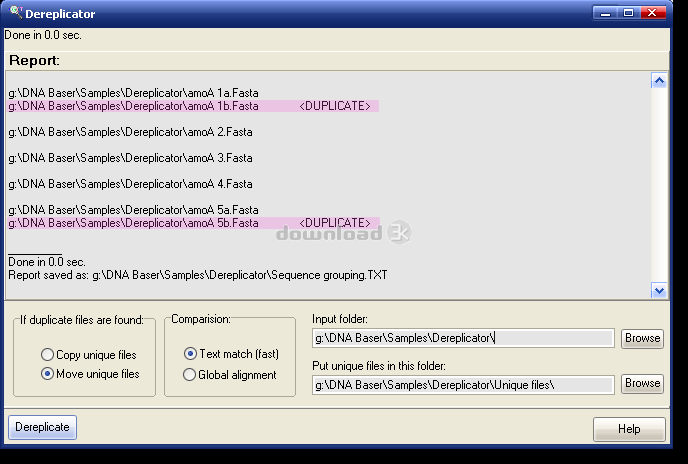
Web site: http://www.DNABaser.comWhen building clone libraries it is often the case that many of the sequenced clones are 100% identical. Therefore, before further DNA sequence analysis, it is necessary to find duplicate DNA sequences and remove them. Our DNA sequence dereplication tool sorts all unique DNA sequences (FASTA) belonging to your clone libraries, by moving/coping them into the specified folder.
Clone Library Dereplicator simplifies the dereplication of all type sequence libraries (16S rRNA, 18S rRNA, 23S rRNA, 28S rRNA, functional and structural proteins) and prepares the raw sequences for subsequent analyses or contig assembly.
DNA Baser Assembler is easy to use software for simple and batch DNA sequence assembly, DNA sequence analysis, contig editing, metadata integration and mutation detection. It also offers a powerful chromatogram viewer/editor. The truly user-friendly interface makes DNA Baser the best choice for DNA contig assembly. For more details, see the DNA BASER Features page.
Why is DNA Baser Assembler special?
Any software company pretends that their product is the best. But let's see for real if DNA Baser can offer you a better proposition. As you will see below we concentrate on adding automatic and batch functions to our product in order to decrease the time. Additional to this, DNA Baser is available at a "kill your competition" price.
Forget about manually trimming the low quality ends of your sequences. DNA Baser Assembler will do it for you!
Do you think you need weeks to assemble hundreds of contigs? What about doing this in minutes? DNA Baser is the only software that can automatically detect and assemble sequences belonging to the same contig based on their filename.
Do you think that is necessary to spend more than 20 minutes to correct discrepancies and mismatches in every contig? Wrong! DNA Baser is the first software which can make correct suggestions in at least 98% of cases.
Have you ever wondered how others laboratories afford to have sequence assembly software in EVERY computer? Simple! They don't spend thousands of dollars for each license. They use DNA Baser Assembler. DNA Baser is affordable, has no annual maintenance fees, technical support is included in price and you have instant access to your key, right after purchase.
You don't have to fill in and submit forms in order to get a trial version. If you want to try it, you can download and install it in less than one minute. No personal data or registration process is required. The trial version is fully functional.
DNA BASER Assembler offers a smart navigation system that takes you to the location of each sequence ambiguity / mutation with a single click.
Requirements: CPU: 333MHz, 64MB RAM, Video 1024x768, 2MB HDD free space
What's new in this version: New: Metadata and batch metadata integration. New: Button to open Windows Explorer in contig's folder, after sequence assembly. New: Remove vectors from single chromatograms. New: SEQUENCE ANALISYS - mark al bases with a confidence level (QV) below a specified threshold, in red. New: Batch convert from chromatogram to Fasta with vector removal and automatic metadata integration. New: Resizable chromatograms. Full support for low quality sample ends editing. 100% compatible with Mac via Parallels/Bootcamp/VMWare. Improved handling of corrupted/invalid ABI/SCF files. Improved contig editor. Improved file association. Improved 'Assemble to reference'. Improved log window. Improved file handling: Before starting the contig, check if all files in the JobList are valid. Invalid samples are automatically removed from Job List so the assembly process can continue without human intervention. Build a list of invalid files and report it. Improved user interface: new toolbar, improved embedded help, interactive help, workflow... Improved sample viewer: 'Mark as trusted/un-trusted' can now be used also in Sample viewer window New: Show error message while trying to open empty/invalid FASTA files Improved: Correctly handle multiple contigs resulted when assembling to a reference. Improved: menu 'Save as Fasta/Seq/Scf' was replaced with 'Save all as...' and 'Save selected as...'. Now the user can choose where to save the file.
Antivirus information
-
Avira:
Clean -
Kaspersky:
Clean -
NOD32:
Clean

Popular downloads in MP3 Audio Video
-
 Realtek High Definition Audio Driver for 2000/XP/2003 32/64-bit R2.74
Realtek High Definition Audio Driver for 2000/XP/2003 32/64-bit R2.74
High definition audio driver from Realtek. -
 3GP Player 2013 1.4
3GP Player 2013 1.4
Free 3GP Player for Pc -
 iTunes 12.13.7.1
iTunes 12.13.7.1
Manage and play your music collection. -
 Windows Media Player 11
Windows Media Player 11
One of the best media player -
 Total Video Converter 3.72
Total Video Converter 3.72
Total Video Converter -
 TVexe TV HD 6.0
TVexe TV HD 6.0
Watch free live TV on your PC now+ Radio -
 K-Lite Codec Pack Full 18.9.0
K-Lite Codec Pack Full 18.9.0
A collection of codecs and related tools. -
 Windows Media Player 9 Codecs Pack
Windows Media Player 9 Codecs Pack
The latest Windows Media codecs -
 VLC media player 3.0.21
VLC media player 3.0.21
A portable app of VLC media player. -
 KMPlayer 4.2.3.21 x86 / 2025.1.21.12 x64
KMPlayer 4.2.3.21 x86 / 2025.1.21.12 x64
A movie and audio player.
-
OS
Win95,Win98,WinME,WinNT 3.x,WinNT 4.x,WinXP,Windows2000,Unix,Linux,Mac PPC -
License
Freeware -
Updated
7/23/2021 -
Downloads
322 (1 last week) -
Size
3.50 MB -
MD5
e1764bef6be7b10b45085041ead79baf -
SHA1
2ab36be11079e1ffed5da33f25ce551cfc79d222 -
Developer
Heracle BioSoft